Why a language-model expert’s intuitions misfire
DNA is the most beguiling analogy in all of machine learning. It is a string. It is written in a tiny alphabet. You read it left to right. It has motifs that look like words, genes that look like sentences, and a “grammar” that biologists have spent a century annotating. If you have trained transformers on text, the leap to genomics feels like a short one — same architecture, new corpus.
It is not a short one. The architectures really do carry over (transformers, state-space models, masked-language-model pre-training, increasingly genomic “foundation models”), which is exactly what makes the analogy dangerous: it hides everything that is different. This post is a field guide for machine-learning scientists moving from natural language into genomics and transcriptomics. What changes is not the network. It is the statistics of the signal, the meaning of a “token,” the fact that the entire species shares essentially one sequence, the biology you must encode to read a single variant, and — the part that quietly sinks most projects — the molecule you can cheaply measure is not the molecule that actually does anything.
A running theme, the mirror image of the one I used for radiology: some things here are genuinely easier than language, and a few are catastrophically harder. (For the imaging counterpart of this argument, see Radiology AI Is Not Computer Vision.) Knowing which is which is the difference between a model that tops a benchmark and one that says something true about biology.
The alphabet is tiny — and stranger than text
Start with the surface, because the surface is where the false comfort lives.
Line up like with like before drawing the comparison. The right counterpart to DNA’s four letters — \(\{A, C, G, T\}\), with RNA swapping \(T\) for \(U\) — is not an NLP tokenizer’s 32,000–100,000-token vocabulary but the 26 letters of written English: both are the raw character set from which everything else is assembled. On that axis DNA’s alphabet is merely small, not exotic. The comparison only gets interesting one level up, at the word — and there the genome’s closest analogue is the gene, of which humans have only ~20,000 (Section 7), each one hundreds to thousands of letters long rather than five. So a four-letter alphabet “sounds like a gift,” and for sheer per-symbol modeling capacity it is — but the comfort is misplaced, because the difficulty was never in the alphabet. Three wrinkles make even the alphabet less clean than it looks.
It is not really four symbols. Real sequence files are littered with N, the
“any base” placeholder for positions the sequencer could not call, and with the
full IUPAC ambiguity set (R = A or G, Y = C or T, W, S, K, M, and so
on) for positions known to be one of a subset. On top of that, reference genomes
use case to encode a second channel: lowercase acgt marks soft-masked
regions — repeats and low-complexity sequence flagged by tools like RepeatMasker —
while uppercase marks the rest. So a and A are the same base carrying
different metadata. A tokenizer that uppercases everything silently discards a
curated annotation; one that treats a and A as distinct tokens doubles the
alphabet for the wrong reason. Neither is obviously right, and the choice is yours
to make consciously.
The hard axis is length, not alphabet. This inverts the usual NLP scaling axis: there is almost nothing to learn about the alphabet; all the difficulty is in the length. The haploid human genome is about \(3.2 \times 10^{9}\) base pairs — a single “document” of 3.2 billion characters, two to three orders of magnitude longer than the entire training context of a long-context LLM. This is why tokenization is a live research question in genomics in a way it is not for English: single-nucleotide tokens give you faithful resolution but punishing sequence lengths; \(k\)-mer tokens (e.g. 6-mers) or byte-pair encodings shorten the sequence but blur the single-base substitutions that often are the signal. The thing you most want to detect — a one-letter change — is the thing aggressive tokenization destroys.
The string has a symmetry text does not. DNA is double-stranded, and the two
strands carry the same information in complementary, reverse-ordered form: the
reverse complement of 5'-GATTACA-3' is 5'-TGTAATC-3'. A gene can live on
either strand, so a motif and its reverse complement are often biologically
equivalent — an equivariance with no analogue in natural language, where reading a
sentence backwards through a letter-substitution cipher is gibberish. Good genomic
models build in reverse-complement equivariance (or augment with it); naive ones
waste capacity relearning each motif twice. Relatedly, strand bias — the two
strands accruing mutations or being sequenced at different rates — is a real
artifact you must model, not a nuisance you can normalize away.
Almost every genome is the same genome
Here is the single biggest statistical difference from a text corpus, and it runs exactly opposite to intuition. Two English documents pulled at random share almost nothing at the token level. Two human genomes are 99.9% identical.
Pairwise nucleotide diversity in humans is about \(\pi \approx 0.001\): roughly one site in a thousand differs between any two people. Across \(3.2 \times 10^{9}\) bases, a typical genome carries on the order of 4–5 million sites that differ from the reference — which sounds like a lot until you remember it is \(0.1\%\) of the sequence. The other \(99.9\%\) is, position for position, the same book. Figure 1a puts this on a log axis against the divergence you would see between species (\(\sim 1.2\%\) human–chimpanzee) and against text, where two unrelated documents differ at essentially every token.
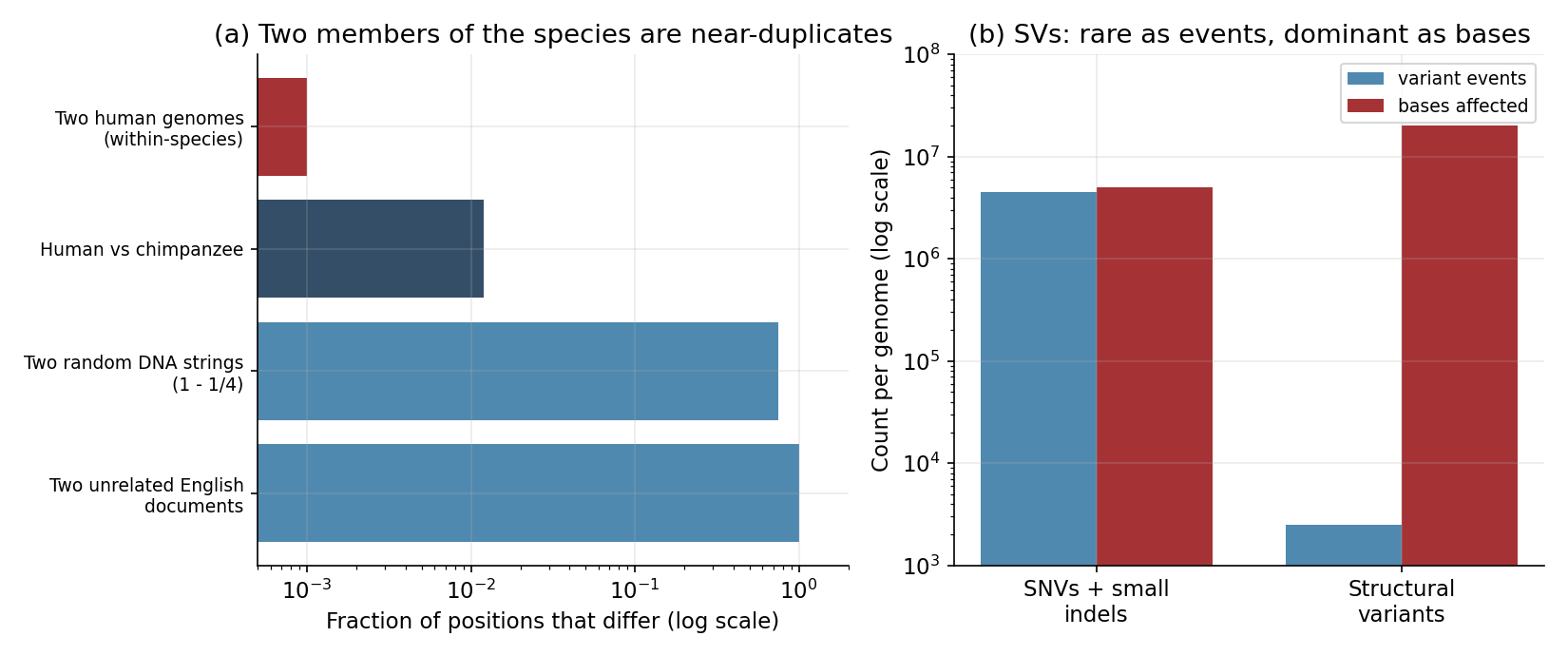
Why so uniform? Human genetic variation is shallow because the population that gave rise to everyone alive passed through a long period of small effective population size (\(N_e\) on the order of \(10^{4}\)) and a series of out-of-Africa bottlenecks tens of thousands of years ago (conventionally placed \(\sim 50{,}000\text{–}70{,}000\) years ago). The practical consequence for a modeler is profound: the common variants you see are old and shared. They are ancestral polymorphisms that predate the bottleneck, inherited by everyone and merely reshuffled into new combinations by recombination each generation. There is not a fresh, independent draw of variation per person; there is one ancestral deck, dealt and re-dealt. On top of that shared deck, each newborn carries only dozens of brand-new mutations — about 70 de novo single-nucleotide variants per generation, from a per-base mutation rate near \(1.2 \times 10^{-8}\). So a person is: the ancestral common variants (shuffled) \(+\) a small private set of rare and de novo ones.
There is a second subtlety in which differences you are counting (Figure 1b). Count variation by events and single-nucleotide variants (SNVs) and small indels dominate — millions of them. Count it by bases affected and the picture flips: a typical genome harbors only a couple of thousand structural variants (deletions, duplications, inversions, insertions of mobile elements), but those rearrange roughly 20 Mb of sequence — far more nucleotide content than all the SNVs combined. Structural variants are simultaneously the largest source of differing bases and the least studied, because they are hard to call from short reads and awkward to represent against a single linear reference. Much of the “missing” signal in genomics lives in exactly the variation our tooling sees worst.
For an ML scientist, the redundancy is not a curiosity — it is a data-leakage hazard that dwarfs anything in NLP:
- Your training and test “documents” are near-identical by construction. A random train/test split over individuals leaves the two sets sharing \(99.9\%\) of their sequence and the overwhelming majority of their common variants. A model can score brilliantly by memorizing ancestral haplotypes that appear on both sides of the split.
- Relatedness and population structure are confounders, not noise. Cryptic relatives, or simply two people of shared ancestry, share long haplotype blocks. This is the genomic analogue of a radiology model learning the scanner instead of the disease: here the model learns ancestry and launders it as signal. Split by family and, where the question demands it, by population; correct for structure with principal components or mixed models; and never trust an evaluation that could be won by recognizing where someone’s grandparents were born.
- The reference itself is a bias. Mapping everyone to one linear reference genome systematically mishandles the variants — especially structural ones — that the reference happens not to contain. Pangenome graph references exist to fight this; ignoring it bakes a population-specific blind spot into your inputs.
Most of the genome is unread — and the labels need a wet lab or a patient
In a text corpus, every token means something to a competent human reader; meaning is in distribution to the annotator. Genomics is not like this. Only about \(1\)–\(2\%\) of the human genome codes for protein. The rest is introns, regulatory elements, structural and repetitive sequence, and vast stretches whose function is genuinely unknown. The ENCODE project assigned “biochemical activity” to most of the genome, but biochemical activity is not the same as function, and the fraction of the genome whose role we can actually read off remains small. Most of the book is written in a language we have only begun to decode.
This reshapes what “supervised learning” can even mean, because the labels are the bottleneck, just as annotation is in radiology — and for a deeper reason than cost:
- Function is not in the sequence the way meaning is in the text. You cannot look at a 200-base enhancer and read its effect the way you can read a sentence. Establishing what a non-coding region does requires a wet-lab experiment — a massively parallel reporter assay, a CRISPR perturbation screen, a knockout — or a population-scale association to a phenotype. The label lives in an experiment or in a patient, not in the characters.
- “Ground truth” is often officially uncertain. Clinical variant databases enshrine this honesty: a large share of catalogued variants are Variants of Uncertain Significance (VUS) — we have seen them but cannot say whether they cause disease. Pathogenicity calls follow formal guidelines (ACMG/AMP), yet different labs reach conflicting classifications for the same variant often enough that reconciling them is its own field. If your training labels are pathogenic/benign calls, you are inheriting both the biology’s uncertainty and the curators’ disagreement — the genomic version of inter-reader variability.
- You cannot eyeball the data. An NLP engineer can sanity-check a labeling pipeline by reading examples. Almost no one can look at a stretch of intron and tell whether a splice-site annotation is right. As in radiology, you need a biologist in the loop continuously, because the data-cleaning decisions (which transcripts to keep, how to treat a multi-mapping read, what counts as “expressed”) are biological judgments wearing a data-engineering disguise.
Meaning travels: cis, trans, and the limits of a context window
The defining structural fact of language modeling over the last few years has been the expanding context window — from a few thousand tokens to a million and beyond — on the premise that if a dependency exists, a long enough window will span it. Genomics tempts you to apply the same logic, and to a point it works: the state-of-the-art regulatory models read enormous windows. Enformer takes about \(200\,\mathrm{kb}\) of sequence; DeepMind’s 2025 AlphaGenome ingests up to \(\sim 1\,\mathrm{Mb}\) — a literal one-million-base context window — to predict regulatory activity. The parallel to long-context LLMs is exact, and deliberate.
But a linear window, however long, runs into a wall that has no NLP analogue, because genomic regulation is not confined to a line:
- Cis-regulation is long-range but at least on-chromosome. Enhancers routinely act over hundreds of kilobases, skipping past nearer genes to their true targets. A \(1\,\mathrm{Mb}\) window is a real attempt to capture this — and it captures a lot of it.
- The genome is folded in 3D. Promoters and enhancers are brought into contact by chromatin looping within topologically associating domains. Two elements far apart on the sequence can be physically adjacent in the nucleus. Linear distance in your input is not regulatory distance in the cell.
- Trans-regulation breaks the line entirely. A transcription factor encoded on chromosome 1 diffuses through the nucleus and binds targets on every chromosome. Trans-eQTLs — variants that affect the expression of genes far away, often on other chromosomes — are exactly this. No sliding window over a single locus, of any length, can see a regulator that lives on a different chromosome and acts through a protein intermediate that is not in the input sequence at all.
That last clause is the crux. In language, the relevant context is always more text; a bigger window is the right tool. In genomics, the relevant context is frequently a diffusible molecule, a 3D contact, or a cell-state variable that the DNA sequence does not contain. The state that determines what a sequence does is not fully written in the sequence. Stretching the context window from \(200\,\mathrm{kb}\) to \(1\,\mathrm{Mb}\) is genuine progress on the cis problem and buys nothing on the trans problem. Be precise about which one your model is actually solving.
You cannot read the data without biology
This is the section a language modeler is most tempted to skip and least able to afford skipping. A handful of biological facts are not background color; they change what a model must represent to be correct.
The reading frame and the genetic code. Protein-coding sequence is read in non-overlapping triplets (codons). With four letters, there are \(4^3 = 64\) codons mapping onto 20 amino acids plus stop — a degenerate code, so several codons specify the same amino acid (most often differing in the third “wobble” position). Three consequences follow immediately: the code is frame-dependent (an insertion or deletion not divisible by three causes a frameshift that garbles everything downstream); the same protein can be written many ways; and a model operating on raw nucleotides has to learn a triplet structure that is given, not discovered.
Silent does not mean neutral. Because the code is degenerate, a single-base change can be synonymous (“silent”) — it leaves the amino acid unchanged. The naive inference is that synonymous variants do not matter. They often do: they can alter codon-usage and translation efficiency, mRNA folding and stability, and — crucially — they can create or destroy splice signals. The hierarchy a model should encode is synonymous / missense / nonsense, but with the explicit caveat that “synonymous” is a statement about the protein sequence, not about function.
Splicing and RNA processing. The path from gene to message is not a copy. A pre-mRNA is spliced — introns removed, exons joined — then capped at the \(5'\) end and polyadenylated at the \(3'\). A variant deep inside an intron, far from any coding base, can create a cryptic splice site and ruin a protein; a variant at an exon boundary can cause exon skipping. This is why “distance to the nearest coding base” is a terrible proxy for “importance,” and why models that ignore splicing miss an entire mechanism of disease.
Driver versus passenger. In cancer genomics the problem is explicitly a signal-detection one. A tumor genome accumulates thousands of somatic mutations, the vast majority of which are passengers — along for the ride, biologically inert. A handful are drivers that actually confer growth advantage. Distinguishing the few drivers from the many passengers, against a mutational background that varies across the genome, is the central inference task — the genomic needle-in-a-haystack.
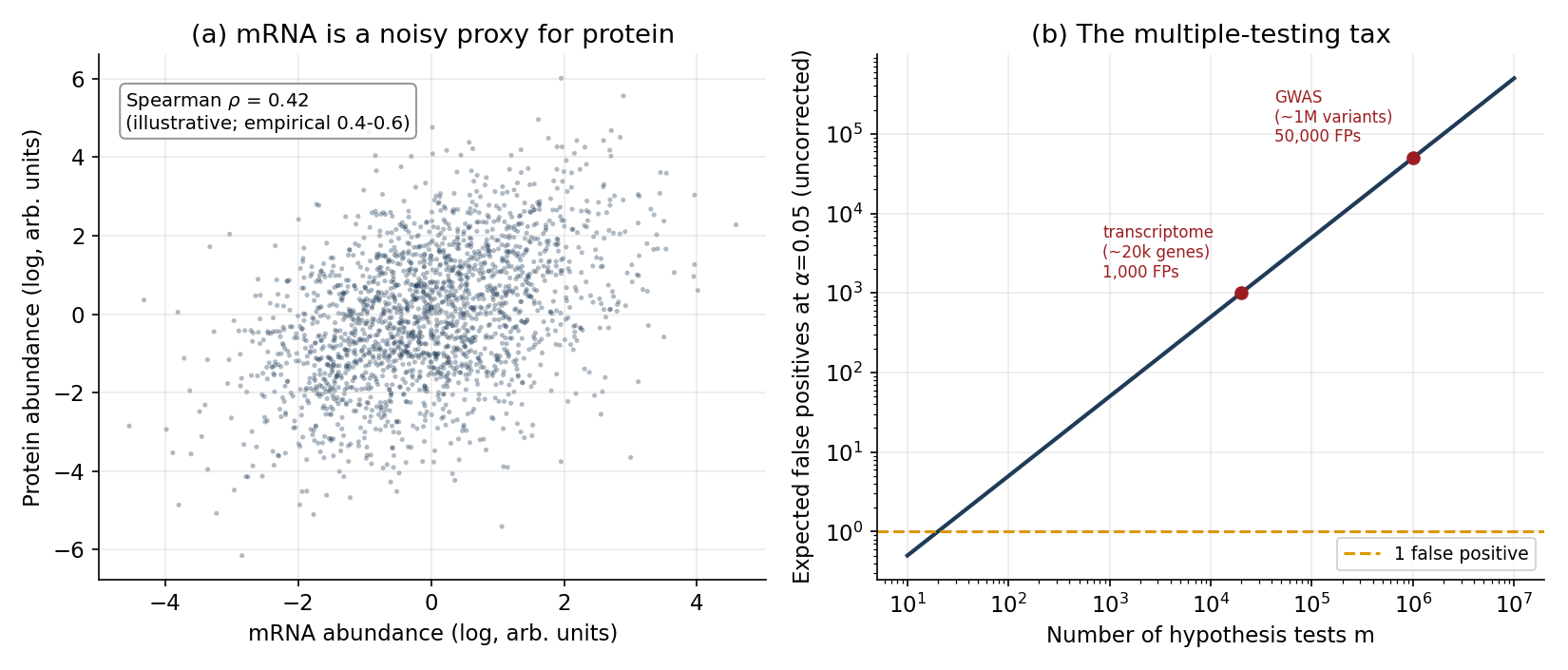
Multiple testing is not optional. When you test millions of variants for association with a trait, or tens of thousands of genes for differential expression, the number of hypotheses is so large that uncorrected \(p\)-values are meaningless. This is why genome-wide association studies adopted a genome-wide significance threshold of \(p < 5 \times 10^{-8}\) — essentially a Bonferroni correction for the \(\sim 10^{6}\) independent common-variant tests across the genome. We return to the arithmetic in Figure 3b; for now, internalize that a “significant” hit at \(p = 10^{-3}\) is, genome-wide, almost certainly noise.
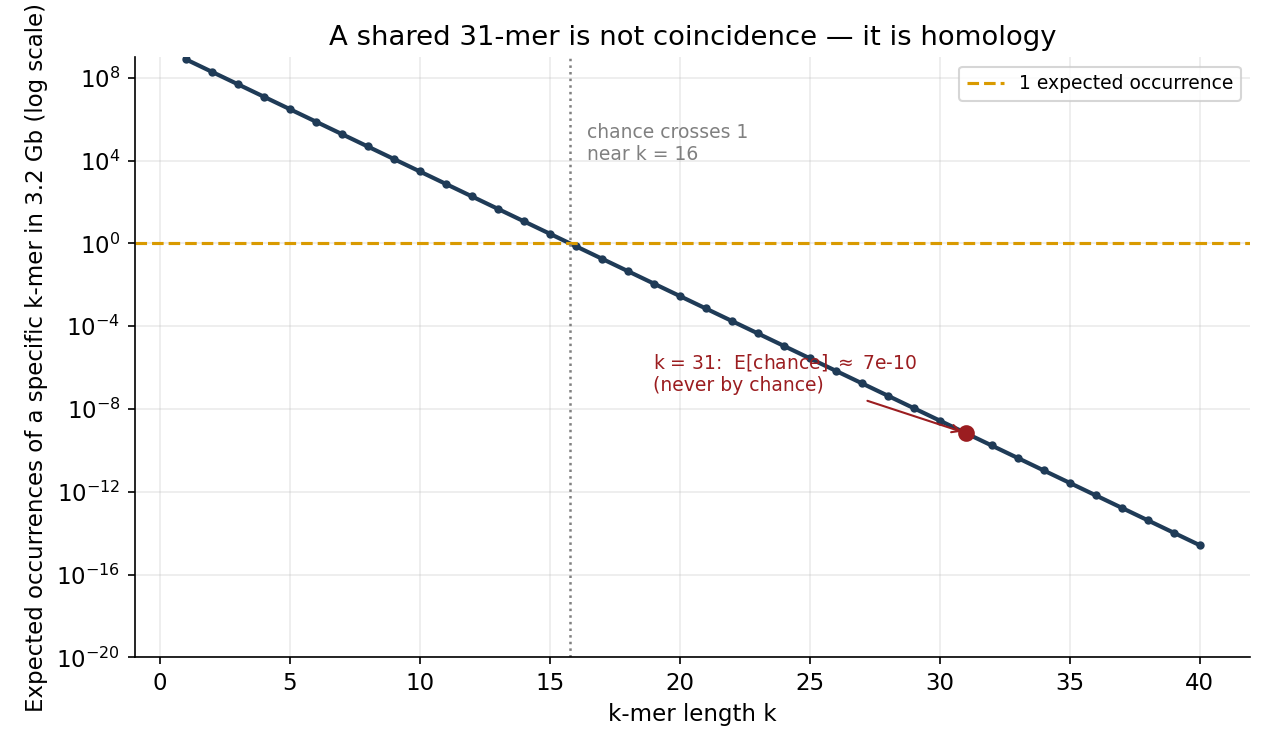
Sequence similarity runs far above chance — and that is the whole point. Here is the calculation that should reframe how a string modeler thinks about DNA. Under a naive model of i.i.d. uniform bases, a specific \(k\)-mer is expected to occur \(G \cdot 4^{-k}\) times in a genome of \(G\) bases:
\[\mathbb{E}[\text{occurrences}] = G \cdot 4^{-k}.\]For \(G = 3.2 \times 10^{9}\), this crosses \(1\) near \(k \approx 16\) and collapses fast (Figure 2). At the \(k = 31\) that bioinformatics tooling routinely uses for exact matching, the expected number of chance occurrences of a given 31-mer is
\[3.2\times 10^{9} \cdot 4^{-31} \approx 7 \times 10^{-10},\]i.e. effectively never. And yet conserved 31-mers are shared constantly — between two people, between human and mouse, across hundreds of millions of years of divergence. The naive random-sequence model predicts these shared long \(k\)-mers should not exist; they exist anyway, by a factor of a billion. That gap is biology: purifying selection conserving functional sequence, preserved RNA secondary structure constraining which substitutions are tolerated, conserved amino-acid motifs (with synonymous wobble underneath), and repetitive elements copied across the genome. The lesson is that string coincidence is the wrong null. When two sequences match more than chance allows, that excess is the signal — homology, conservation, selection — and a model that treats DNA as a random string will systematically misread it.
The unit problem: long genes, many transcripts, slippery semantics
In language the semantic unit is convenient: a word is a few characters, a sentence a few dozen words, and meaning is reasonably local and human-readable. The genomic “word” is nothing like this.
Genes are long, and their meaning is delocalized. A protein-coding sequence is typically on the order of \(1\)–\(2\,\mathrm{kb}\) (encoding a protein of very roughly \(\sim 375\) amino acids, with wide spread across the proteome), but the gene — exons plus the introns between them — frequently spans tens to hundreds of kilobases; dystrophin spans about \(2.2\,\mathrm{Mb}\). The information that specifies one protein is scattered across a huge genomic interval, interrupted by introns, and its realized “meaning” depends on cell type, developmental stage, and regulatory state. Compared with a five-letter English word whose meaning is right there on the page, the semantics of a gene are spread out, context-dependent, and much harder to capture in a fixed embedding.
One gene is many messages. It is common shorthand that humans have “about 20,000 genes” — and the protein-coding count, \(\sim 19{,}900\text{–}20{,}000\) in GENCODE, is indeed remarkably small. But that number badly understates the functional vocabulary, because alternative splicing lets a single gene produce many distinct transcripts (isoforms). GENCODE annotates well over \(200{,}000\) transcripts — an order of magnitude more than genes — and a single gene can yield dozens of isoforms with different, sometimes opposing, functions. So the mapping from “gene” to “thing that acts” is one-to-many, and a transcriptomic model that collapses expression to the gene level is averaging over functionally distinct products. The right unit is frequently the transcript, not the gene — and transcript-level labels are scarcer and noisier.
The molecule you sequence is not the molecule that acts
Now the deepest mismatch, and the one most likely to invalidate a confident conclusion. Proteins do the work of the cell — they catalyze, signal, transport, and build. DNA is the blueprint and RNA the working copy, but the actors are proteins. And yet the overwhelming majority of “expression” data, and nearly all of the trendy single-cell atlases, measure RNA, not protein. We routinely study the script and infer the performance.
That inference is shakier than the field’s habits suggest. Across many careful studies, the correlation between a gene’s mRNA level and its protein level is moderate at best — typically a Spearman \(\rho\) in the \(0.4\)–\(0.6\) range, and lower still when you look at changes over time rather than steady-state across genes. Schwanhäusser and colleagues found mRNA explained well under half the variance in protein abundance; Vogel and Marcotte, Liu, Beyer and Aebersold, and Buccitelli and Selbach all converge on the same message — translation rates, protein half-lives, and post-translational regulation drive a large share of protein levels that mRNA simply does not see. Edfors and colleagues showed the relationship is gene-specific: each gene has roughly its own mRNA-to-protein conversion factor, so a single global model is wrong per gene. Figure 3a illustrates the consequence — even at the optimistic end of that range, knowing a gene’s mRNA leaves its protein level uncertain across a wide band.
So why does the field overwhelmingly sequence RNA if protein is what matters? Not because anyone thinks RNA is the better readout — because of technology:
- RNA can be amplified; protein cannot. Reverse transcription plus PCR turns a handful of molecules into a sequenceable library, so RNA-seq reaches single-cell and even single-molecule sensitivity. There is no PCR for proteins — no way to exponentially copy a polypeptide — so mass-spectrometry proteomics works with whatever is in the sample.
- RNA is genome-templated, so we know what to look for. Every transcript maps back to a sequence we can align against a reference. Proteins must be inferred from fragmentary peptide spectra, and the proteome’s enormous dynamic range means abundant proteins drown out the rare ones we often care about most.
- Throughput and cost. RNA-seq is cheap, standardized, and scales to millions of cells; comprehensive single-cell proteomics is still hard, lower-throughput, and far less complete.
The honest framing is the same one that recurs throughout this post: we optimize a convenient proxy. RNA abundance is to protein activity what a radiology label mined from a report is to the underlying pathology — useful, scalable, and systematically wrong in ways you must keep in view. A transcriptomic model that reports “expression” is making a claim about the script; whether the performance followed is a separate, and weaker, inference.
The data: a near-duplicate corpus, batch effects, and who is in it
Genomics is, paradoxically, both data-rich and data-poor. There is an enormous and growing public infrastructure (Table 1), far better than radiology’s. But the redundancy of Section 3, the batch effects below, and the demographics of who has been sequenced mean that effective sample size lags raw counts badly.
Table: Major public genomics / transcriptomics resources. Counts are as reported by the source publications; “variants” and “samples” are not comparable units across rows.
| Resource | What it is | Reported scale | Citation (DOI) |
|---|---|---|---|
| 1000 Genomes | Reference catalogue of human variation | 2,504 individuals, 26 populations; ~88M variants | 10.1038/nature15393 |
| gnomAD | Aggregated exomes + genomes; constraint metrics | 125,748 exomes + 15,708 genomes (v2) | 10.1038/s41586-020-2308-7 |
| UK Biobank | Population cohort, genotype + deep phenotype | ~500,000 participants | 10.1038/s41586-018-0579-z |
| TCGA | Pan-cancer tumor/normal multi-omics | ~11,000 tumors, 33 cancer types | 10.1038/ng.2764 |
| GTEx | Genetic regulation of expression across tissues | 17,382 RNA-seq samples, 54 tissues, 948 donors | 10.1126/science.aaz1776 |
| ENCODE | Functional/regulatory element annotation | Genome-wide assays across many cell types | 10.1038/nature11247 |
| GENCODE | Reference gene/transcript annotation | ~20,000 coding genes; >200,000 transcripts | 10.1093/nar/gkaa1087 |
| Geuvadis | RNA-seq paired to 1000 Genomes genotypes | 462 individuals, 5 populations | 10.1038/nature12531 |
| Tabula Sapiens | Multi-organ single-cell atlas | ~500,000 cells, ~24 tissues | 10.1126/science.abl4896 |
| T2T-CHM13 | First complete (telomere-to-telomere) human genome | 1 gapless assembly | 10.1126/science.abj6987 |
Two structural problems run underneath these numbers.
Batch effects are the scanner-heterogeneity of genomics. A sequencing readout is the end of a long wet-and-dry pipeline, and every stage is a covariate that shifts across labs: library preparation chemistry, sequencing platform (short-read Illumina vs. long-read PacBio/Nanopore), read length and depth, PCR amplification bias, RNA quality (RIN) and degradation, the alignment software, and — easy to forget — the reference build itself (GRCh37 vs. GRCh38 vs. the new T2T-CHM13). Two expression datasets can differ more by batch than by biology, and models readily learn the batch. The discipline that grew up around this — careful normalization, batch-correction methods, mixed models, harmonized pipelines — is the genomic counterpart to vendor-aware augmentation and intensity normalization in imaging. As there, the danger is symmetric: under-correct and you measure the lab; over-correct and you erase the biology.
The corpus is not representative of humanity. A large majority of participants in genome-wide studies are of European ancestry. This is the genomic version of the subgroup-power trap: a polygenic risk score trained predominantly on European- ancestry data transfers poorly to people of other ancestries, because the tag variants, allele frequencies, and linkage structure differ. A model can post excellent aggregate metrics and still be least accurate for the populations most underserved by existing tools. Splitting and evaluating by ancestry, and stating plainly which groups you are and are not powered to serve, is not optional diligence — it is the difference between a fair tool and an inequitable one.
The famous models — and what they do and don’t solve
The reason this analogy is everywhere right now is that the transformer toolkit has produced genuinely landmark genomics results. It is worth knowing the map, and being precise about what each model does and does not address from the list above.
- AlphaFold2 (Jumper et al., 2021) predicts protein 3D structure from amino-acid sequence at near-experimental accuracy — arguably the field’s defining success. Note what it sidesteps: it operates on the protein, taking the molecule that acts as a given, and says nothing about whether or how much of that protein the cell makes.
- Enformer (Avsec et al., 2021) and AlphaGenome (DeepMind, 2025) attack the cis-regulatory problem head-on, predicting expression and chromatin readouts from sequence across \(\sim 200\,\mathrm{kb}\) and up to \(\sim 1\,\mathrm{Mb}\) windows respectively. They are the state of the art on long-range cis effects — and, per Section 5, structurally blind to trans regulation that acts through diffusible proteins or other chromosomes.
- DNABERT (Ji et al., 2021), the Nucleotide Transformer (Dalla-Torre et al., 2024), and Evo (Nguyen et al., 2024) are DNA “language models” — masked or autoregressive pre-training over genomic sequence, transferred to downstream tasks. They inherit, and must confront, every tokenization and redundancy issue in Sections 2–3.
- scGPT (Cui et al., 2024) and Geneformer (Theodoris et al., 2023) bring the foundation-model recipe to single-cell transcriptomics, learning representations of cell state from large RNA-expression atlases — which means they live entirely on the RNA side of the proxy gap in Section 8.
The pattern across the map is the through-line of this post: these models are spectacular within the slice of the problem they address, and it is on the modeler to know which slice that is. AlphaFold takes the protein as input; the regulatory models see only cis; the single-cell models see only RNA. None of that diminishes them — it just means the honest question is never “does the benchmark go up,” but “which part of the biology did this actually capture, and which part is still missing.”
Takeaways
If you remember five things moving from natural language to genomics:
- The alphabet is a trap, not a gift. Four letters (plus
N, IUPAC codes, and case-as-metadata), but the difficulty is the 3.2-billion-character length, the reverse-complement symmetry, and a tokenization choice that can destroy the single-base signal you came for. - The whole species is one near-duplicate corpus. Two genomes differ at \(0.1\%\) of sites; common variants are old and shared, private variation is dozens of mutations, and most differing bases hide in understudied structural variants. Plan your splits around leakage, relatedness, and population structure from day one.
- Most of the genome is unread, and labels live in experiments or patients. Function is not in the sequence the way meaning is in text; ground truth is often an official “uncertain,” and you cannot eyeball it. Keep a biologist in the loop.
- Regulation defeats the context window. A \(1\,\mathrm{Mb}\) window is real progress on cis and no progress on trans: the determining context is often a protein, a 3D contact, or a cell state that the sequence does not contain.
- You are usually modeling a proxy. RNA is not protein, and the correlation is only \(\sim 0.4\)–\(0.6\); “expression” is the script, not the performance. Encode the biology — codons, splicing, silent-but-not-neutral, drivers vs. passengers, multiple testing, similarity-beyond-chance — or your string model will confidently misread the genome.
See the accompanying notebook.ipynb for the redundancy arithmetic, the \(k\)-mer
calculation, the proxy simulation, the multiple-testing counts behind Figures 1–3,
and an automated check that every citation below resolves.
References
- Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi:10.1038/nature15393
- Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. doi:10.1038/nature15394
- Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi:10.1038/s41586-020-2308-7
- Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. doi:10.1038/s41586-018-0579-z
- Weinstein JN, Collisson EA, Mills GB, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. doi:10.1038/ng.2764
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–1330. doi:10.1126/science.aaz1776
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi:10.1038/nature11247
- Frankish A, Diekhans M, Jungreis I, et al. GENCODE 2021. Nucleic Acids Res. 2021;49(D1):D916–D923. doi:10.1093/nar/gkaa1087
- Lappalainen T, Sammeth M, Friedländer MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. doi:10.1038/nature12531
- Tabula Sapiens Consortium. The Tabula Sapiens: a multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376(6594):eabl4896. doi:10.1126/science.abl4896
- Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. Science. 2022;376(6588):44–53. doi:10.1126/science.abj6987
- Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488(7412):471–475. doi:10.1038/nature11396
- Schwanhäusser B, Busse D, Li N, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337–342. doi:10.1038/nature10098
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13(4):227–232. doi:10.1038/nrg3185
- Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell. 2016;165(3):535–550. doi:10.1016/j.cell.2016.03.014
- Edfors F, Danielsson F, Hallström BM, et al. Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol Syst Biol. 2016;12(10):883. doi:10.15252/msb.20167144
- Buccitelli C, Selbach M. mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet. 2020;21(10):630–644. doi:10.1038/s41576-020-0258-4
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. doi:10.1038/s41586-021-03819-2
- Avsec Ž, Agarwal V, Visentin D, et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat Methods. 2021;18(10):1196–1203. doi:10.1038/s41592-021-01252-x
- Avsec Ž, Latysheva N, Cheng J, et al. AlphaGenome: advancing regulatory variant effect prediction with a unified DNA sequence model. bioRxiv. 2025. doi:10.1101/2025.06.25.661532. See also https://deepmind.google/blog/alphagenome-ai-for-better-understanding-the-genome/
- Ji Y, Zhou Z, Liu H, Davuluri RV. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 2021;37(15):2112–2120. doi:10.1093/bioinformatics/btab083
- Dalla-Torre H, Gonzalez L, Mendoza-Revilla J, et al. Nucleotide Transformer: building and evaluating robust foundation models for human genomics. Nat Methods. 2024;22(2):287–297. doi:10.1038/s41592-024-02523-z
- Nguyen E, Poli M, Durrant MG, et al. Sequence modeling and design from molecular to genome scale with Evo. Science. 2024;386(6723):eado9336. doi:10.1126/science.ado9336
- Cui H, Wang C, Maan H, et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat Methods. 2024;21(8):1470–1480. doi:10.1038/s41592-024-02201-0
- Theodoris CV, Xiao L, Chopra A, et al. Transfer learning enables predictions in network biology. Nature. 2023;618(7965):616–624. doi:10.1038/s41586-023-06139-9
Reproduce all analyses in this post here.
]]>